Illumina dominates the HTS market especially when it comes to resequencing for genetic variation. The comparatively lower error rate and high output of Illumina sequencing makes it the top choice for genotyping. For de novo genome sequencing, these short Illumina reads yield less contiguous assemblies but usually of higher quality for subsequent annotation. There are plenty of different Illumina sequencing platforms with more constantly being released or discontinued. I defer to the Illumina website for that as this is comprehensive enough and would really depend on which platform you have access to.
General pipeline
The Illumina libraries are loaded into “lanes” within flowcells. Depending on the sequencing facility, you may be able to purchase just a fraction of a lane, a single lane, or the entire flowcell. Different machines also have different numbers of lanes per flowcell and different costs per flowcell.
Anatomy of an Illumina library
Before we dive into sequencing, we might want to familiarize ourselves with the anatomy of an Illumina library. The steps to create an Illumina library can be followed here. The end product is basically your fragmented template DNA flanked by oligo indexes which are compatible with Illumina sequencing machines.

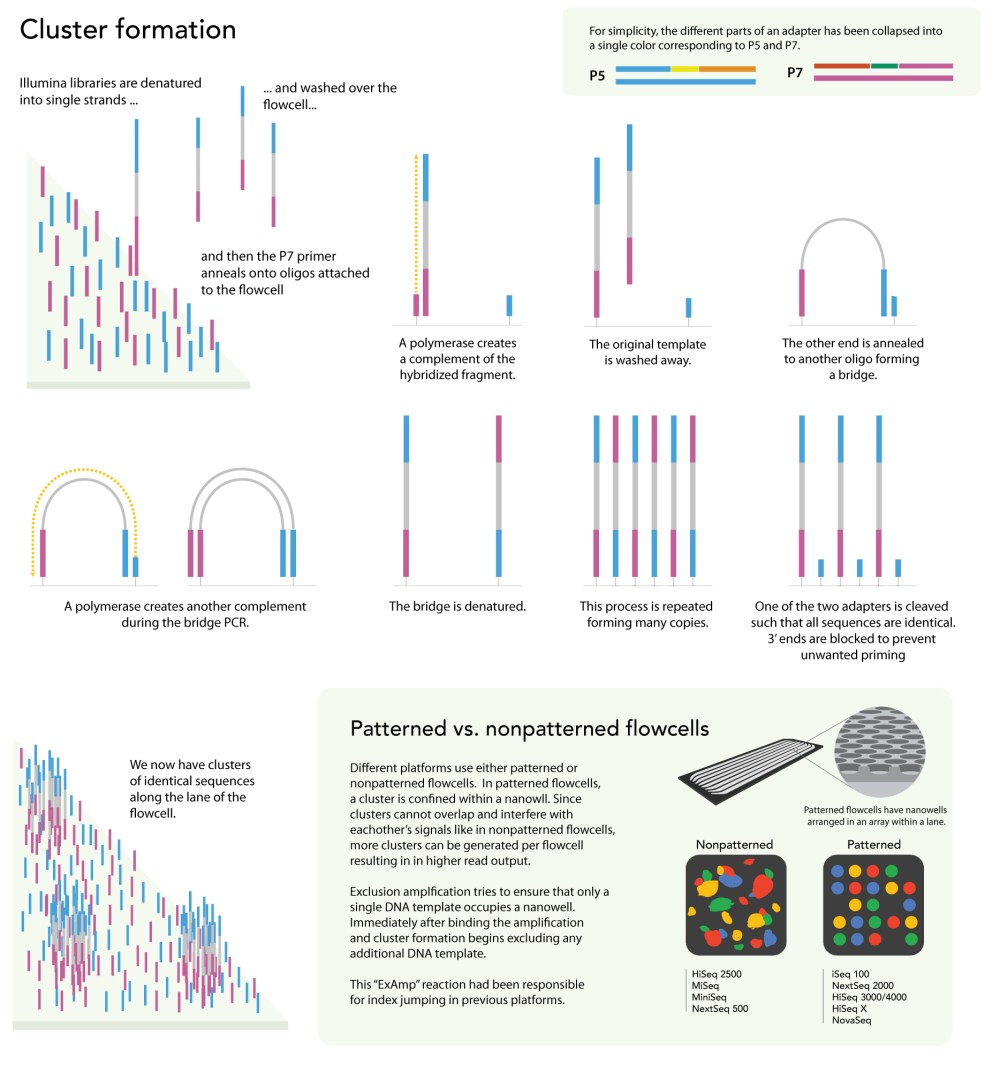
Illumina sequencing
This is simply a static representation of how Illumina sequencing works. Watching this video may be more helpful with understanding the steps. After choosing a specific Illumina platform, you must also choose the type of sequencing you would like to be done. Here are some important terms to know to determine the type of run you would like to do:
- Sequence length – Refers to how many basepairs will be read from your sequences. This ranges from 50, 100, 150, and even 300bp for MiSeq.
- Single-end (SE) vs. paired-end (PE) – SE refers to sequencing only one direction (forward) while PE sequences in both directions (forward and reverse)
- Single-index vs. dual-index – Refers to whether both i5 and i7 indexes contain barcodes which determines if these cycles need to be run.